����һ��MDR����
����2017��5��5�գ��W�˹ٷ��ڿ���Official Journal of the European Union����ʽ�l���˚W���t����е��Ҏ��REGULATION(EU)2017/745�����Q“MDR”����MDR��ȡ��Directives 90/385/EEC����Դֲ����t����еָ�and 93/42/EEC���t����еָ�������MDR Article 123��Ҫ��MDR��2017��5��26����ʽ��Ч�����c2020��5��26������ʽȡ��MDD��93/42/EEC����AIMDD��90/385/EEC����
��������MDR����Ҫ׃��
����1.�U���ˑ��÷���
����2.������µĸ������е�Ķ��x
����3.�������t����е�ķ��
����4.��������е��ͨ�ð�ȫ������Ҫ��
����5.�ӏ������g�ļ���Ҫ��
����6.�ӏ���е���к�ıO��
����7.�����R���u�r���PҪ��
����8.���Eudamed������Ľ�����ʹ��
����9.�����е�Ŀ����ԣ�UDI��
����10.��NB��������Ҫ��
��������MDR�m�÷����U��
������MDR���H������MDD��AIMDD���w�����ЮaƷ��߀���w���T������е���坍��������������е���Լ�Annex XVI���e�ğo�A���t��Ŀ�ĵĮaƷ������ͫ���沿����ע�䡢�y����Ƥ�w���ƺ����ݵȮaƷ��
��������ijЩˎе�Y�ϮaƷ��Ԕ��ՈҊArticle1��8��9����
��������ijЩ�ɷǻ��Ի�̎���ǻ��Ե����Դ�M����������������ض��aƷ��
���������Q�H��������Ŀ�Ļ���һ�N���t��Ŀ�ģ����ڹ��ܺ��L�U��������������t����е���ض��aƷ�M
�������{�ײ�����е����MDR��������Ҫ������������u������
���������l���x��ݗ�����е���t����;��ܛ����
�����ġ�MDR������µĸ������е�Ķ��x
����MDR�������˺ܶ��µĸ����MDD�е�14��������ӵ��F�ڵ�71������������һЩ�R��ԇ������к�O�ܷ���ĸ����“recall,withdraw,serious incident,clinical consent,clinical benefit”.Ԕ��ՈҊArticle2
�����塢��е�ķ��׃��
������MDD��MDR����е�Է֞��Ĵ��I�IIa�IIb�III�
����MDD���c������P��93/42/EEC�е�Annex IX��������ָ��MEDDEV 2.4/1 Rev.9���µ�MDR��Article51��Annex VIIIԔ���U���ˮaƷ�ķ����Ϣ����Ҫ׃������MDD��“18�l”���Ğ�MDR��“22�l”��
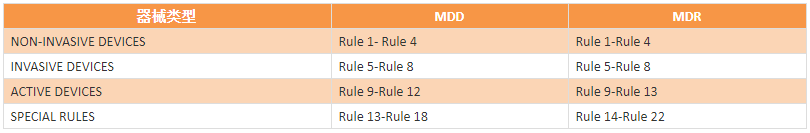
����Rule1-Rule 4��NON-INVASIVE DEVICES����������е
����Rule3�������������w��ֱ�ӏ����w�������̥ȡ���w��ʹ�õ����w�������M������ܣ�Ȼ����ֲ���ע���w�ȣ������е��III�
����Rule 5-Rule 8��INVASIVE DEVICES��������е
����Rule 8����ԭ���Ļ��A�������ˣ���Դֲ����е�������P�������鷿ֲ��������K���a�W���������P���ÓQ�ֱ���c�������|���g�P�ÓQֲ�����III�
����Rule 9-Rule 13��ACTIVE DEVICES��Դ��е
����Rule9����ԭ���Ļ��A��������“ᘌ��ί�Ŀ��ጷ���xݗ�����Դ��е”�Լ�“���ڿ��ơ��O�y��ֱ��Ӱ���Դֲ��ʽ��е”���@�ɴ����е����II b�
����Rule11�������ӣ���������ṩ�\����ί�Ŀ�ěQ����Ϣ�ͱO�y�����^�̵�ܛ��������II a�����ܛ���I�
����Rule 14-Rule 22��SPECIAL RULES����Ҏ�t
����Rule14���Mһ��������“���������wѪҺ��Ѫ�{���t���aƷ”���Ҫ��
����Rule 18���Mһ������“���÷ǻ��Ի�̎���ǻ��Ե����w�����Դ�M�����������������Ƴɵ���е”�ķ��Ҫ��
����Rule19�����ӌ��{�ײ�����е�ķ��Ҫ��
����Rule20��������ͨ�^���뷽ʽ���c���w�����P��������е�ķ��
����Rule 21���������������w���������|�����w�����
����Rule22�������˾��м��ɻ�ϲ��\��ܵ���Դ�ί���е�ķ��
��������߀�h����MDD�Ќ�Ѫ���ĆΪ����
����������е��ͨ�ð�ȫ������Ҫ��
������MDD��Annex 1 ESSENTIAL REQUIREMENTS�Mһ�����ƞ�“ANNEX I GENERAL SAFETY AND PERFORMANCE REQUIREMENTS”����ԭ����13���l�����ӵ��F�ڵ�23����ͬ�rMDD��Article 13:Information supplied by the manufacturer��MDR������һ���Ϊ����¹�“CHAPTER III REQUIREMENTS REGARDING THE INFORMATION SUPPLIED WITH THE DEVICE”�������˶��l����Ҫ���{���L�U��������؞�����OӋ�����a���N�ۡ����к�O�ܵ������aƷ�����С�
�����ߡ����g�ļ���Ҫ��
����MDR�������ˌ����g�ļ����ݵ�Ҫ�������_ָ�����к�O��Ӌ���Ͱ�ȫ�Ը���棨PSUR�����Ǽ��g�ļ���һ���֣���Ҫ���������к�O���wϵ�ռ����Y�ό����g�ļ���������Ϣ�M�и��¡�
�����ˡ����g�ļ��Ļ�������
������е�f���c����ָ��
��������׃�ͺ���������е�f���c����ָ�ˣ��Լ����õ�ǰ���������е����Ϣ��
�����������ṩ����Ϣ
�����OӋ�c������Ϣ
����ͨ�ð�ȫ�c����Ҫ��
������������ϸ��I�ṩ��ͨ�ð�ȫ�c����Ҫ����C���Y�ϡ�
�����L�U����������L�U����
�����aƷ��C�c�_�J
�����R��ǰ���R�������������R���u�rӋ��/��棬PMCFӋ��/��棩���Լ�ᘌ���ˎ��е�����w/�����Դ�M�������������Ƃ����е���������w����������е�����Мy��������е�ȵ����P������Ϣ
�����š����к�O�ܵļ��g�ļ�
����AnnexIII TECHNICAL DOCUMENTATION ON POST-MARKET SURVEILLANCEԔ���f����Ҫ����Article83-86�������к�O�ܵ��ļ����������к�O��Ӌ�������к�O�܈����ڰ�ȫ�Ը���棨PSUR����
����ʮ�����������ļ�
����ANNEX IV EU DECLARATION OF CONFORMITYԔ���f����“��������”�ļ������ă��ݡ�
����ʮһ���ӏ���е���к�O���wϵ
����Chapter VII POST-MARKET SURVEILLANCE,VIGILANCE AND MARKET SURVEILLANCE�����f�����к�O�ܡ�������Ј��O�ܡ�
������������ʩ�;S�o���к�O���wϵ��ҊArticle83����
�������{���к�O���wϵ؞�������������ڣ���������¡�
��������“���к�O��Ӌ��”��ҊArticle84�������w����ҊAnnex III��
����I���е����“���к�O�܈��”��ҊArticle85����
����IIa��IIb��III���е����“���ڰ�ȫ�Ը���棨PSUR��”��ҊArticle86����
����PSUR�趨�ڸ��²����鼼�g�ļ���һ���֡�
����������������к�O�����ϵ�y��ҊArticle 92����
������������еʹ�É������g��������ʩPMCF��ȡ�õ��R���������R���u�r�����g�ļ��M�и��£�Annex XIV part B����
����ʮ���������R���u�r���PҪ��
�����·�Ҏ�����
����Ҫ�����Article61���XIV partA���С��u�����������R���u�r�Y�ϣ�
����������ض�III�IIb���е��CER��Ҫ���]��ԃ����С�M����Ҋ��
������ֲ���III���е��������]�R���о���
����Ҫ��CER����PMCFȡ�Ô����M�и��£�
����ᘌ�IIIͿ�ֲ����е�������CER���µ��l�ʣ�
�������_�C�����|��ͬ���迼�]�����c��
����Ҫ�����c�L�U�����������
����ʮ����Eudamed������
�����·�Ҏ�����
�������_�W���t����е�����죨Eudamed������Ŀ�ĺͰ�������Ϣ��Article 33����
������Ϣ�Ĺ��_�ԣ�
����Ҫ��III���е��ֲ��ʽ��е����ȫ���R��������Ϣͨ�^Eudamed���_�š�
����ʮ�ġ������е�Ŀ����ԣ�UDI��
���������ƺ��о���е�⣬������е���轨��UDIϵ�y��
����UDI��Ϣ�w�F�ژ˺�����b�ϣ����������b�䣩��
����UDI-DI��Ϣ��Ҫ�d���ڷ��������У�ҊArticle27����
����Annex VI Part B���UDI-DI��������Ϣ��
������ֲ�롢�؏�ʹ�á�ܛ������������е��UDI������Ҫ��ҊAnnex VI Part C��
�������b��˺���UDI��ʩ�ĕr�gҊArticle123(f)��
����UDI�l�Ќ��w�ɚW��ί�T��ָ����
�����^���ԣ�Article 120ָ��“��ί�T��������27(2)�lָ���l�Ќ��wǰ��GS1��HIBCC��ICCBBA����ҕ��ָ���İl�Ќ��w”��
����ʮ�塢��NB����ć���Ҫ��
������“����C��”���°�MDR���M��ƪ�������M���ܺ�Ҫ���M����������NB�谴�ո��VII��Ҫ��������ՈMDR���������˵��ڙ�
����ʮ��������׃��
����EU߀��һ����ʹ����е������̎����Article17���ͽo���ߵ�ֲ����еֲ���│��Article18�������Ҫ��

 ��һ�ӹ��̎���
��һ�ӹ��̎���